Materials and Methods
Trial Design
The “Evaluation of Pimobendan In dogs with Cardiomegaly caused by preclinical mitral valve disease” (EPIC) trial was a prospective multicenter, blinded, randomized, placebo-controlled study. The trial protocol was prepared by independent cardiologists (AB, JH, and SG) in conjunction with the sponsor and was approved by an ethical review committee at each site where this was required. The contract between the sponsor and lead investigators (AB, JH, and SG) stipulated that the latter would have full access to all results and the right to independently publish, regardless of trial outcome.
Dogs
Enrollment Criteria.
Dogs were eligible for participation in the study provided that the owner had given informed consent.
To be eligible for inclusion, a dog had to be 6 years of age or older, have a body weight ≥4.1 and ≤15 kg, have a characteristic systolic heart murmur of moderate to high intensity (≥ grade 3/613) with maximal intensity over the mitral area, have echocardiographic evidence of advanced MMVD defined as characteristic valvular lesions of the mitral valve apparatus, MR on the color Doppler echocardiogram, and have echocardiographic evidence of left atrial and left ventricular dilatation, defined as a left atrial-to-aortic root ratio (LA/Ao)14 ≥ 1.6 and body weight normalized left ventricular internal diameter in diastole (LVIDDN)15 ≥ 1.7, in addition to radiographic evidence of cardiomegaly (vertebral heart sum (VHS) > 10.5).16
Exclusion Criteria.
Dogs were excluded from the study if they had any of the following: known clinically important systemic or other organ-related disease that was expected to limit the dog’s life expectancy or required chronic cardiovascular medication precluded as part of the trial (Table 1). Dogs with hypothyroidism could be included provided the investigator deemed them clinically stable on treatment. Dogs with current or previous evidence of cardiogenic pulmonary edema, pulmonary venous congestion or both, cardiac disease other than MMVD, clinically significant supraventricular, ventricular tachyarrhythmias or both (i.e, requiring antiarrhythmic treatment), or evidence of pulmonary hypertension considered to be clinically relevant (RV:RA pressure gradient > 65 mmHg) were excluded. Dogs with a history of chronic or recent administration (>14 days of duration or within 30 days of intended enrollment) of any medication listed in Table 1 were excluded. Dogs that were pregnant or lactating were not eligible for enrollment. In the event that before study enrollment, a dog had received short-term treatment (<14 days) with agents listed in Table 1, but was no longer receiving treatment and had not received it within 30 days of intended enrollment, then the dog was eligible for inclusion.

Table 1 – Prohibited cardiovascular agents. Chronic administration of these agents before study entry was an exclusion criterion. Initiation of any of these agents during the study was considered an event in the time-to-first-event analysis. Initiation of chronic treatment resulted in a dog being censored from the per-protocol population.
Study Sites
Dogs were recruited by investigators specializing in veterinary cardiology at 36 centers: 18 in the United States, 3 in the UK, 3 in Germany, 3 in France, 2 in Japan, 2 in Sweden, 1 in each of Italy, Spain, the Netherlands, Canada, and Australia. (All authors except one (PW) were investigators recruiting cases).
Randomization and Allocation
Block randomization17 was used with a 1:1 allocation ratio in blocks of 20 to maintain similar sample sizes in both treatment groups. The randomization sequence was generated by the manufacturer of the trial medicationa by computer software.b Each investigator was initially assigned 10 consecutive case numbers from a block of 20, ensuring that each investigator did not know how many cases assigned to each treatment were under their care.
When an investigator recruited a new dog, that dog was assigned the next available case number and received the preassigned treatment. During recruitment, some case numbers were reallocated between investigators in order to reach the recruitment target. Case numbers reallocated to an investigator were always from a treatment block different to that from which the investigator’s original 10 case numbers came. The maximum allowable number of cases enrolled by any single center was 20.
Blinding
Investigators, owners, study monitors, the statisticianc , and the sponsor were blinded to treatment allocation.
The blinding code for the treatment groups was held by individuals who had no other role in the study. Predefined procedures were available to permit unblinding of individual cases in the event of a medical emergency. Unblinding could be achieved by contacting named individuals who held the randomization list; they could then break the treatment code and inform the investigator of the treatment the animal was receiving. In the event of unblinding, a dog would be censored from the study at the time of unblinding.
Neither the investigators, nor the monitor, nor the sponsor of the study had access to the randomization list.
Trial Medication
Pimobendan verum (Vetmedin 2.5 mg chewable tablets) was administered PO at a target dose of 0.4–0.6 mg/kg/d as per registered label instructions in most countries where the product was licensed. The calculated daily dose was divided into 2 administrations and adjusted to a suitable number of tablets. Placebo was administered PO according to the calculated daily dose for pimobendan verum tablets, divided into 2 administrations, and adjusted to a suitable number of placebo tablets. Placebo and verum tablets and packaging containers were visually identical.
Dogs were dosed in the morning and evening approximately 12 hours apart. Dose of study medication was not adjusted throughout the study.
Populations Analyzed
Any dog that was randomized and received at least 1 dose of study medication was included in the intention-to-treat (ITT) population. Any dog in the ITT population that was confirmed to have met all inclusion criteria (and none of the exclusion criteria) was included in the per-protocol (PP) population until one of the following occurred: the dog reached the primary endpoint, the dog was censored from the primary endpoint analysis due to the occurrence of an event that precluded continuation in that population, or the end of the study was reached.
Concomitant Treatments
All concomitant medications that dogs were receiving at the time of enrollment or received during the course of the study were recorded.
A dog was censored from the per-protocol (PP) primary endpoint analysis if an attending veterinarian deemed it necessary to chronically administer open-label cardiovascular medication(s) for an indication other than having reached the primary endpoint. A list of such cardiovascular medications (Table 1) was prospectively defined and included in the protocol. An exception to this rule was that short-term use of medications in Table 1 was allowed for a period of <5 days if a dog required anesthesia or sedation, or in the event that a dog enrolled in the study was inadvertently administered one of the medications listed in Table 1 for a period of <3 days, but the reason for administration was not substantiated and chronic medication was unnecessary. Medications, the chronic administration of which did not result in a dog being censored from the PP analysis of the primary endpoint, were predefined in the protocol and are listed in Table 2. Initiation of chronic treatment with any medication in Table 1 or Table 2 was recorded as an event. All commercially available topical treatments for ears and eyes were allowed even if some ingredients are found in Tables 1 and 2, and their chronic administration was not considered an event nor did they preclude enrollment in the study.

Table 2 – Agents that dogs were allowed receive while remaining in the per-protocol population. Initiation of any of these agents during the study was considered an event in the time-to-first-event analysis.
Schedule of Events
Before inclusion, a case history was taken for each dog. The dog then underwent a physical examination, measurement of systolic arterial blood pressure (SAP), echocardiography, thoracic radiography, and routine hematology and blood biochemistry (performed at laboratories local to each investigator) with a minimum database consisting of packed cell volume (PCV), ALT (GPT), total protein, creatinine, potassium (K+), and sodium (Na+) concentrations. Re-examinations were scheduled at day 35, and approximately 4 months after inclusion. Thereafter, the dogs were scheduled for re-examination every 4 months. Dogs enrolled to the study underwent appropriate clinical monitoring including home monitoring of respiratory rate.
A dog that reached the primary endpoint and survived was treated with open-label pimobendan and other cardiovascular medications at the discretion of the investigator. All dogs in the ITT population were followed until they died, were euthanized, were lost to follow-up, or the end of the study was reached. The date of death, if it was known to have occurred, was recorded.
Clinical Evaluation
At inclusion, dog characteristics such as breed, age, sex, and neutering status were documented. The body weight, body condition score (BCS)d, and rectal temperature were measured.
Quality-of-Life Observations After history taking and clinical examination, the following variables were scored (according to the system outlined in Table 3): appetite, demeanor, exercise tolerance, coughing, nocturnal dyspnea/cough, and fainting.
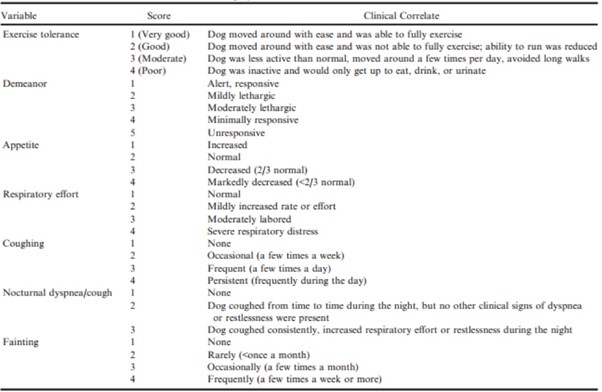
Table 3 – Ordinal scoring system for clinical variables recorded at baseline
Circulatory and Respiratory Variables
The respiratory rate was measured. Cardiac auscultation was performed to detect the presence of any arrhythmia, and to measure the heart rate and grade heart murmur intensity on a scale of 1–6.13 Systolic arterial blood pressure was measured before inclusion by 1 of 2 methods: Doppler sphygmomanometry or oscillometry.
Echocardiography
Echocardiography was performed on unsedated dogs. The following measurements were taken from at least 3 cardiac cycles and the mean was recorded: the LA/Ao obtained from the right parasternal short-axis 2D view as previously described,14 the left ventricular internal diameter at end diastole (LVIDd), and left ventricular internal diameter at end systole (LVIDs) measured on the M-mode echocardiogram, obtained from the right parasternal short-axis view.18 M-mode values were used to derive the fractional shortening (FS%). Normalized dimensions15 were calculated according to the following formulae:
normalized LVIDd (LVIDDN) = LVIDd(cm)/(BW (kg))0.294;
normalized LVIDs (LVIDSN) = LVIDs(cm)/(BW (kg))0.315.
Thoracic Radiography
Thoracic radiography was performed at inclusion and at the time a dog was considered to have developed signs of CHF. Right lateral and dorsoventral projections were used to evaluate the thorax. Cardiac size was assessed by the VHS method16 and pulmonary edema and congestion were recorded, when considered to be present, by the attending cardiologist.
Primary Endpoint
The primary endpoint was a composite of the development of left-sided CHF, or euthanasia for a cardiac reason, or death presumed to be cardiac in origin in dogs in the PP population. The primary outcome variable of the study was the time period from inclusion until the primary endpoint was reached.
A dog was considered to have left-sided CHF when there was radiographic evidence of cardiogenic pulmonary edema as indicated by an interstitial or alveolar lung pattern. In addition to these radiographic findings, the dog must have been showing contemporaneous clinical signs consistent with left-sided CHF including increased respiratory effort and rate by comparison with previously noted values for this patient.
Two members of an endpoint committee, comprised of the 3 lead investigators (AB, JH, and SG) and 2 additional investigators (RSt and GW), reviewed the radiographs and case records for each dog reaching the CHF component of the primary endpoint in order to verify that the endpoint had been reached. In the event of a disagreement between the 2 members of the endpoint committee, the case was adjudicated by a third endpoint committee member.
Endpoint committee members did not evaluate their own cases and were blinded to treatment allocation at the time of adjudication.
If a dog died in the absence of evidence of a noncardiac cause of death before radiographic confirmation of pulmonary edema, it was also considered to have experienced cardiac death and therefore reached the primary endpoint.
Dogs that reached the primary endpoint and in which no relevant protocol deviation or violations occurred during the study were included in the PP analysis. Dogs in which a major protocol deviation occurred during the study (e.g, the owner was not compliant with study procedures, or there were lengthy treatment gaps comprising a single period without trial medication of >30 days or the total duration of period without medication was >10% of the dog’s total time on study) were included in the PP analysis until the time when the protocol deviation occurred, at which time they were censored. However, these dogs could still be eligible for inclusion in the ITT analyses.
At any point in the study, an owner or investigator could remove a dog from the study for reasons of animal welfare, suspected adverse drug reaction, and illness or injury that prohibited the dog’s continuation.
Secondary Endpoints
A dog was considered to have experienced an event if it reached the primary endpoint, underwent euthanasia or died for a noncardiac reason, had chronic medication initiated (Table 1 or Table 2), had a CHF endpoint that was not verified by the endpoint committee, the owner became noncompliant with study procedures, experienced lengthy treatment gaps, or the dog was withdrawn from the study by the owner or investigator. The time to the first of these events experienced by dogs in the PP population was analyzed as a prospectively defined secondary endpoint. (For further information, see Table S2).
Time to death by any cause was also a prospectively defined secondary endpoint and analyzed in the ITT population.
Appropriate pharmacovigilance was undertaken and adverse events experienced by dogs on the study were noted, classified, and reported in accordance with applicable guidelines.
Data Management
Data management was undertaken by an independent data management company.e All clinical and dispenser records were collected from all centers. Data were verified and double entered into a database by separate individuals. After data entry, the 2 sets of data were compared to verify accuracy of entry of data and any discrepancies between the 2 databases were explored and resolved.
Blinding was maintained during data entry and audit. All decisions on censoring and exclusions from the study were made on the basis of predetermined criteria by the members of the lead committee who remained blinded.
Statistical Methods
Power Calculation
Power calculations for study population size were performed by nQuery platform.f The planned duration of the accrual period was 2 years and the maximum duration of followup was 5 years. A minimum of 3 years follow-up was planned after the accrual period. Results of prior studies had shown that the median time to the onset of left-sided CHF in dogs with stage B2 MMVD is approximately 27 months.6,7 Based on these assumptions, 150 animals per group were considered necessary to detect a difference in median time to the onset of left-sided CHF of greater than or equal to 13 months with a power of 80% and an alpha of 5%. To compensate for possible dropouts, 180 animals per group were included in each treatment group.
Populations Analyzed
In both the interim and final analyses, all efficacy analyses with respect to the primary endpoint were planned as analyses comparing treatment groups in the PP population. Time to first event was also compared between treatment groups in the PP population. The safety analyses, all-cause mortality, and proportion of suspected serious or worse adverse events were evaluated in the ITT population.
Interim Analysis
A preplanned interim analysis was undertaken with predefined stopping criteria for convincing evidence of efficacy and safety, performed on data obtained after 80% of the initial anticipated study period was complete. Unblinding and termination of the study only occurred if deemed necessary by the data interim evaluation committee according to prespecified criteria (Figure S1). The committee consisted of 3 independent (to the study) persons: 1 biostatistician and 2 experts in canine cardiology. The P-value for stopping on the basis of convincing evidence of efficacy with respect to the primary endpoint was decided by appropriate statistical softwaref and set at P < .01477.
Final Analysis
Descriptive statistics for continuous variables are reported as median values with interquartile range (IQR); for categorical and ordinal variables, they are reported as frequency and proportions. For the analysis of the final data, each of the variables obtained at baseline was assessed for homogeneity between treatment groups in the PP populations. All continuous and ordinal baseline variables were compared between groups by a Mann-Whitney-Wilcoxon test. For categorical data, a chi-square or Fisher’s exact test was used. No adjustment was made for multiple comparisons. The P-value for the final analysis was corrected according to the O’Brien-Fleming alpha spending function.19 The critical two-sided nominal efficacy alpha level for the final analysis was calculated to be 0.04551 by appropriate statistical software.g The local alpha levels were calculated for a total type I error level of 5% with a power of 80 % and the assumptions of a hazard ratio of 0.667 and proportional hazards.
In time-to-event analyses, a log-rank test with right censoring was used to determine whether a significant difference existed between the 2 treatment groups in the probability of the event of interest occurring at any time point. The Kaplan-Meier method was used to estimate the median time to endpoint for each treatment group and plot time to event curves.
Univariate Cox proportional hazards analysis with right censoring was performed for the effect of treatment and each baseline variable, to determine whether there was any association with time to primary endpoint. For the purpose of the Cox proportional hazards analysis, categories of ordinal baseline variables with a small number of observations (<5 observations) at a single level were combined with the adjacent level to create a larger group with the same directional tendency. The robustness of the treatment effect was evaluated by adjusted univariable Cox proportional hazards analysis to determine the influence of each baseline variable individually on the treatment hazard ratio. Multivariable Cox proportional hazards analysis was performed in a backward stepwise manner. Baseline variables were selected for entry to the model if they had a P-value of <.2 in the univariate analysis. Variables entered into the multivariable analysis were assessed for multicollinearity and to ensure that the proportional hazards assumption was met. The variable with the highest P-value was eliminated at each subsequent step, with reanalysis between steps, until the final model was obtained with all the remaining variables having a P-value <.05. A second exploratory multivariable model was created as above but excluding echocardiographic variables in the first iteration, and also run in a backward stepwise manner until all variables in the final model had a P-value <.05. Firstorder interaction terms between each pair of variables remaining in the final model were evaluated and included in each model if found to be significant. Proportions of dogs that experienced severe or worse adverse events were compared between groups by a chi-square test.
For all analyses other than those specifically mentioned above, an alpha of 5% was used. All analyses were two-tailed. Statistical analyses were performed by a statisticianc independent of the sponsor by a commercially available software program.h In preparation of the manuscript, authors followed the recommendations given in the CONSORT statement for reporting randomized clinical trials.20
Results
Recruitment to the study began in October 2010 and finished in June 2013. Follow-up was continued until the study was closed on the first of March 2015. The interim analysis was conducted in January 2015 on data collected up to October 1, 2014, in which the criteria for unblinding and stopping the study were met with respect to the time to primary endpoint.
The statistician and 1 lead investigator (AB) were therefore unblinded at this time. All other lead investigators (JH and SG) and those involved in data management remained blinded until data entry was complete.
Three hundred and sixty dogs were enrolled in the study and randomized to receive trial medication. The median number of dogs recruited per center was 9 (range 1–20). One dog was allocated a case number but did not complete enrollment and never received trial medication. The outcome for the remaining 359 dogs in the ITT population is summarized in Figure 1. After randomization, 4 dogs randomized to placebo and 1 dog randomized to pimobendan were excluded because they were subsequently found to have failed an inclusion criterion (4 dogs did not meet at least one of the heart size criteria and 1 dog had been chronically pretreated before enrollment to the study). The remaining 354 dogs—178 receiving pimobendan and 176 receiving placebo—comprised the PP population. The PP population consisted of 161 (45.5%) Cavalier King Charles Spaniels (CKCS), 21 (5.9%) Dachshunds, 13 (3.7%) Miniature Schnauzers, 8 (2.3%) each of Poodles and Yorkshire Terriers, 98 (27.7%) other pure breeds, and 45 (12.7%) mixed breeds. The median age of the enrolled population was 9.0 years (IQR 7–11)
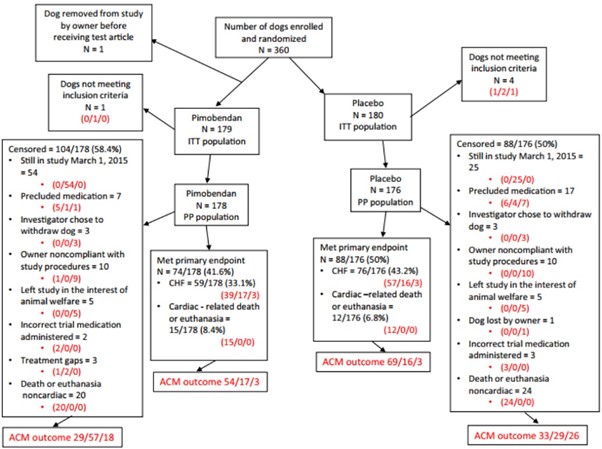
Figure 1 – A flow chart indicating the outcome of 360 dogs randomized in the study. Where 3 numbers appear in red in the diagram, they represent the outcome of dogs in each subgroup with respect to the all-cause mortality analysis. They indicate, in the order in which they appear, dogs known to have died/dogs known to be alive/dogs lost to follow-up. ACM, all-cause mortality, ITT; intention to treat; PP, per protocol; CHF, congestive heart failure.
Baseline characteristics of the 2 groups were summarized and compared (Table 4). The groups were balanced for all baseline variables with the exception of breed and BCS. In the PP population of dogs receiving pimobendan, the median dose received was 0.49 mg/kg/d (IQR 0.441–0.528) divided into 2 doses.
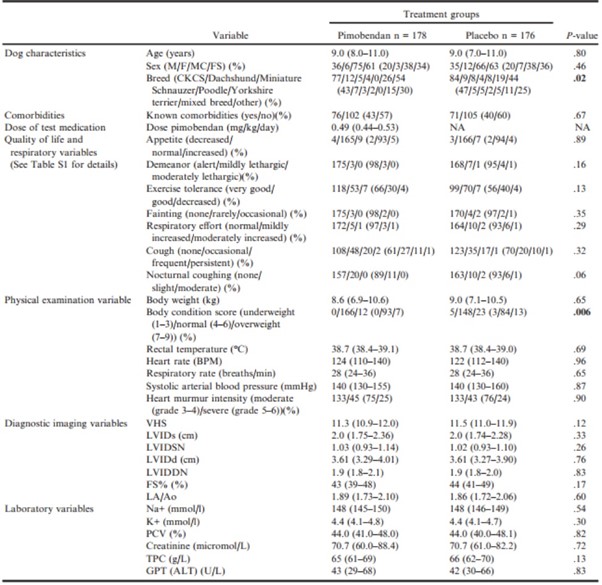
Table 4 – Baseline characteristics of the 2 treatment groups in the per-protocol population and their comparison.
Continuous variables are reported as median (interquartile range). Categorical variables are reported as number (%).
ALT, alanine aminotransferase; BCS, body condition score; BPM, beats per minute; CKCS, Cavalier King Charles Spaniels; F, female; FS, female spayed; FS%, fractional shortening; GPT, glutamic pyruvate transaminase; K+, potassium concentration; LA/Ao, left atrial-toaortic root ratio; LVIDd, left ventricular internal diameter in diastole; LVIDDN, normalized left ventricular internal diameter in diastole; LVIDs, left ventricular internal diameter in systole; LVIDSN, normalized left ventricular internal diameter in systole; M, male; MN, male
neutered; Na+, sodium concentration; PCV, packed cell volume; TPC, total protein concentration; VHS, vertebral heart sum. P-values that appear in bold are < 0.05.
The median time in study for the PP population was 623 days (IQR 292–959). One hundred and sixty-two of the 354 dogs reached the primary endpoint giving an overall event rate of 45.8%. The median time in study for all dogs that reached the primary endpoint was 433.5 days (IQR 242-718). One hundred and ninety-two dogs were censored from the analysis of the primary endpoint, including 104 dogs receiving pimobendan and 88 dogs receiving placebo. Of these, 79 dogs (54 dogs receiving pimobendan and 25 dogs receiving placebo) were still alive, not having reached the primary endpoint, at the end of the study. The median time in study for these dogs was 1056 days (IQR 976-1238). The cause of censoring for the remaining 113 dogs is shown in Figure 1. The median time in study for these 113 dogs was 502 days (IQR 228–787). The proportion of dogs reaching the primary endpoint was not different between groups (pimobendan, 74/178 (41.6%); placebo, 88/176 (50.0%); P = .14).
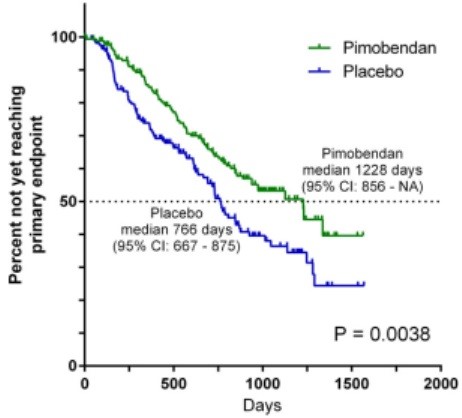
Figure 2 – Kaplan-Meier survival curves plotting the estimated percentage of dogs in each group in the per-protocol population that have not yet met the primary endpoint (congestive heart failure or cardiac death), against time. There were 178 dogs in the pimobendan group and 176 dogs in the placebo group at the outset. CI, confidence interval; NA, not able to calculate.
The estimated median time to the primary endpoint was 1228 days (95% confidence intervals (CI): 856–NA) in the pimobendan group and 766 days in the placebo group (95% CI: 667–875) (Fig 2). The risk of a dog in the pimobendan group reaching the primary endpoint, at any time point, was lower (P = .0038) and the hazard ratio was 0.64 (95% CI: 0.47–0.87) compared with a dog in the placebo group. The times taken for dogs in the 2 treatment groups to reach the individual components of the composite primary endpoint were compared and are summarized in Table 5. The proportions of dogs in each group experiencing each of the 3 individual components of the primary endpoint, CHF, spontaneous cardiac death, and cardiac euthanasia, were not different between groups (P = .063, P = .134, and P = .218).
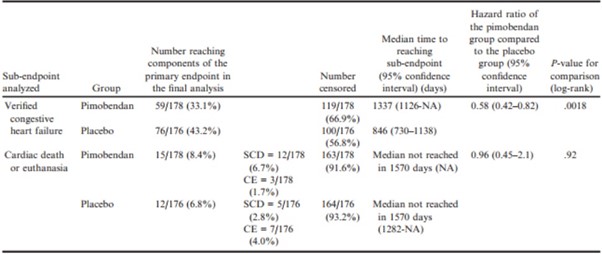
Table 5 – Subanalyses of the primary endpoint.
CE, cardiac euthanasia; NA, Not able to calculate; SCD, spontaneous cardiac death.
Nine dogs initially thought to have developed CHF did not have the presence of radiographic signs of CHF verified by the endpoint committee. Three of these dogs were in the pimobendan group and 6 in the placebo group. Failure to confirm CHF was either due to radiographs being present but failing to demonstrate evidence of an interstitial/alveolar lung pattern, or radiographs not having been obtained at the time of the CHF event. In 8 of these cases, medications listed in Table 1 were administered and these dogs were therefore censored at the time of the suspected heart failure for receiving precluded medication. In 1 case, severe clinical signs resulted in euthanasia of the patient before radiographic confirmation of CHF and this was subsequently (before unblinding) reclassified as a cardiac-related euthanasia.
In the time-to-first-event analysis, 130 dogs in the pimobendan group and 158 dogs in the placebo group experienced an event. Forty-eight dogs in the pimobendan group and 18 dogs in the placebo group were censored in this analysis. The median time to the first event was 640 (95% CI: 555–753) days in the pimobendan group versus 406 (95% CI: 316–527) days in the placebo group (P < .0001) (Fig 3). The number of first events attributable to the introduction of medications from Tables 1 and 2 and their reasons for introduction are summarized according to treatment group in Table 6.
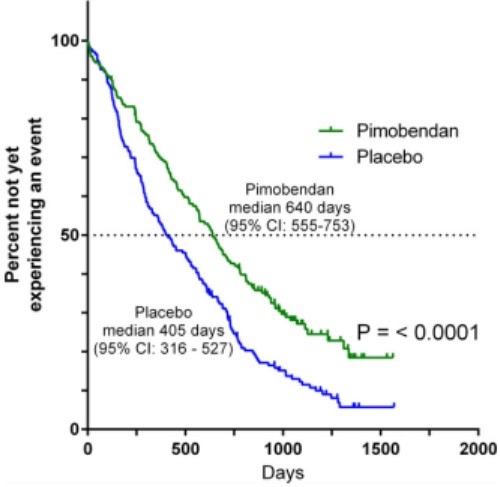
Figure 3 – Kaplan-Meier survival curves plotting the estimated percentage of dogs in each group in the per-protocol population that have not yet experienced an event (defined as having reached the primary endpoint, undergone euthanasia or died for a noncardiac reason, had chronic medication initiated (Table 1 or Table 2), had a congestive heart failure endpoint that was not verified by the endpoint committee, the owner became noncompliant with study procedures, or the dog was withdrawn from the study by the owner or investigator), against time. There were 178 dogs in the pimobendan group and 176 dogs in the placebo group at the outset. CI, confidence interval.
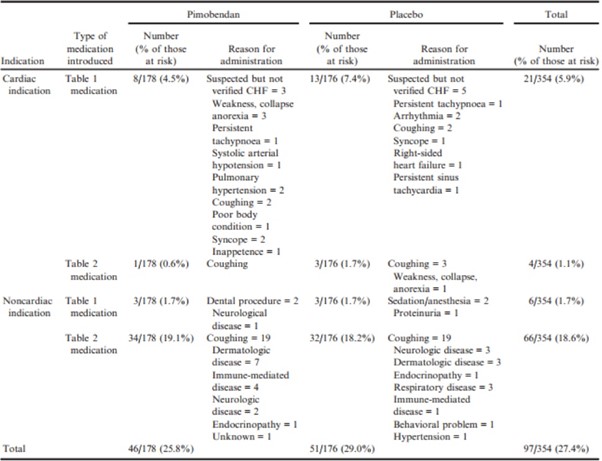
Table 6 – Numbers of dogs for which administration of medication represented the first event in the “time to first event” including the type of medication administered and the reasons given for initiating medication. Note that more than 1 reason could be given for administration of medication and therefore the numbers in the “reason for administration” may not add up to the total in the adjacent column.
The hazard ratio for the treatment effect, when adjusted for the individual effect of each of the 32 baseline variables, did not change substantially (range 0.575–0.665), and the 95% confidence intervals of the estimated hazard ratio never included the value 1 (Figure S2). In the univariate Cox proportional hazards analysis, in addition to treatment, of the 32 baseline variables tested, the following 16 variables demonstrated an association with the time to primary endpoint at P < .2 and were entered in the first iteration of the multivariable analysis: Miniature Schnauzer (yes/no), appetite, exercise tolerance, fainting, respiratory effort, cough, nocturnal coughing/dyspnea, respiratory rate, heart rate, SAP, VHS, FS%, LVIDDN, LA/Ao, PCV, and creatinine. The final model of the multivariable analysis is summarized in Table 7 (and Figure S3).
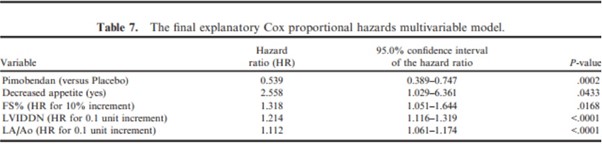
FS%, fractional shortening; HR, hazard ratio; LA/Ao, left atrial-to-aortic root ratio; LVIDDN, normalized left ventricular internal
diameter in diastole.
The final multivariable model without inclusion of echocardiographic measurements is detailed in Table 8.
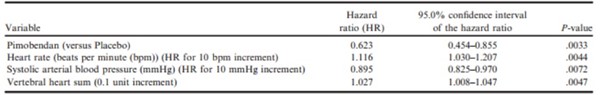
Table 8 – The final exploratory Cox proportional hazards multivariable model without echocardiographic indices.
With respect to the safety analyses in the ITT population, there were no significant differences between groups in the type and severity of adverse events experienced. The proportion of dogs in each group experiencing adverse events during the study was not different (P = .82) (Table 9).
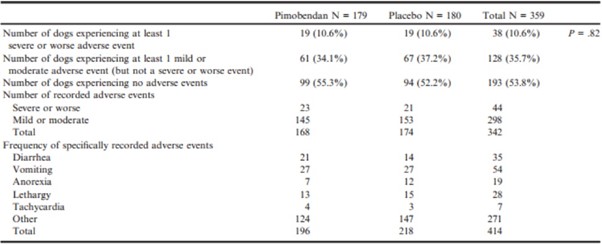
Table 9 – The nature and severity of adverse events experienced by the dogs in the 2 treatment groups during the study. In the course of the study, dogs could experience more than 1 adverse event or experience more than 1 clinical sign at the time of an adverse event.
One hundred and eighty-six dogs were known to have died by any cause at the date of study closure, 83 of 179 dogs (46.4%) in the pimobendan group and 103 of 180 dogs (57.2%) in the placebo group (P = .0260). The median time to death by all causes was 1059 (95% CI: 952–NA) days in the pimobendan group and 902 (95% CI: 747–1061) days in the placebo group (P = .012) (Figure S4).